The use of Good Laboratory Practices (GLP) has become an essential component of scientific research, but its origins can be traced all the way back to a scandal in 1978.
The convictions of the executives at IBT Labs led to the development of GLP regulations, which have since become a cornerstone of quality testing and development.
Read on to learn more about the history of GLP and its importance in modern research.
History of Good Laboratory Practices (GLP)
Formal rules for good laboratory practices began in the US in 1978, when a scandal at an industrial product safety testing lab resulted in the establishment of quality assurance principles that remain fundamental to scientific research today.
Once widely used by the government and industry to test thousands of new chemicals, such as pharmaceuticals, pesticides, and others for safety, Industrial Bio-Test Laboratories (IBT Labs) operated the largest facility of its kind and performed more than one-third of all toxicology testing in the United States. But the convictions of the company’s president and several top executives in 1983 for extensive scientific misconduct, fraud and falsifying statements, would lead to reforms in the regulation of pesticides in the United States and Canada and the proposal and development of Good Laboratory Practices (GLP). These practices became an evaluative series of record-keeping protocols designed to help prevent fraud, promote the generation of high-quality data, and ensure data validity. Today, GLP regulations have become a basic expectation of laboratories across the country.
“IBT once was considered one of the nation's most reliable authorities for determining potential cancer and other hazards from new chemicals and compounds,” the Washington Post reported. “The investigation forced the Environmental Protection Agency and dozens of chemical companies that had used IBT's test reports to retest hundreds of new substances to determine if they were safe for humans.”
“Testimony from former IBT workers during the six 1/2-month trial indicated that rats and mice used in the complex dosage-and-examination sequences escaped from their cages and were replaced by other animals from different test groups,” The Washington Post continued, “and that other rodents died in abnormally high numbers that went unreported. One witness recalled that the cages for the animals were filthy, their doors left open and that animals died of thirst or drowned in their water troughs.”
In 1978, the FDA issued the Guidance for Industry Good Laboratory Practices Regulations in response to the scandal at IBT-Labs.
According to Laura N. Vandenberg, in her 2021 book, Advances in Pharmacology, with the GLP regulations, the FDA sought to “ensure that laboratories producing data for regulatory purposes have a specific organizational structure, that they follow established scientific procedures and that data are recorded in a way that protects integrity and ensures traceability.”
How far have we come with GLP
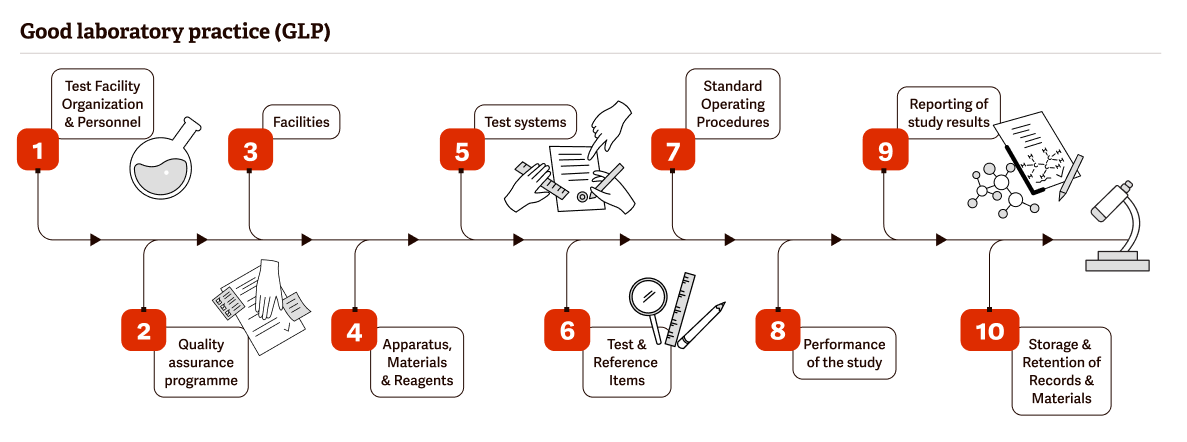
Today, GLP regulations have become a cornerstone of quality testing and development.
According to the Quality Assurance Journal, “the FDA requires nonclinical studies of new drugs, food additives, and chemicals to predict their safety and potential efficacy in humans. The significance of the information gained from these studies requires that they be conducted according to sound scientific principles and with strict attention to quality assurance and quality control. Human health and safety are dependent upon the decisions made from these studies.”
What happens if laboratories are GLP non-compliant?
Does FDA reject nonclinical laboratory studies that have not been conducted in full compliance with the GLPs? According to the questions and answers addressed by the U.S. Department of Health and Human Services (HHS) FDA Office of Regulatory Affairs, not necessarily. “The GLP Compliance Program provides guidance on the issue. For FDA to reject a study, it is necessary to find that there were deviations from the GLPs and that these deviations were of such a nature as to compromise the quality and integrity of the study covered by the agency inspection.”
GLPs for nonclinical laboratory studies are not required, and labs are not certified by the FDA. However, without GLPs, there is little chance of FDA approval of products resulting from those studies. Research in the US not conducted under GLPs or research done outside the US not conducted according to the Organization for Economic Co-operation and Development (OECD) Guidelines (or FDA rules) might be inadmissible in support of a New Drug Application (NDA) in the US. The requirements relate to how laboratory studies are conducted rather than the research itself.
Vandenberg states in “Advances in Pharmacology” that overall, the GLP regulations were “proposed to ensure that laboratories producing data for regulatory purposes have a specific organizational structure, that they follow established scientific procedures, and that data are recorded in a way that protects integrity and ensures traceability.”
Distinct from the test guidelines utilized by any laboratory, Vandenberg writes that “becoming a GLP certified laboratory, and maintaining GLP certification, is a lengthy and expensive endeavor.” Studies conducted according to GLP guidelines can cost 30–100% more than the same studies that are not GLP compliant, she writes.
Although the term GLP is most often associated with the pharmaceutical industry and the required nonclinical animal testing before approval of new drug products, it also applies to many other non-pharmaceutical agents such as medical devices and biologics.
“These regulations are intended to assure the quality and integrity of the data necessary to support FDA investigational new drug (IND) and investigational device exemption (IDE) applications for first-in-human investigations, as well as for subsequent FDA marketing approval,” she continues, adding that the regulations are not solely a US-initiative. “The US GLP regulations are not the only GLP regulations that regulators use worldwide. Many other countries, including the European Union, follow the OECD Principles of Good Laboratory Practice (GLP).”
Del W. Huntsinger of BASF Corporation wrote in a PubMed article that since the inception of the FDA GLP regulations in 1979, the OECD Principles of GLP in 1981, and the finalization of the EPA GLP program in 1983, “there have been recognizable differences among the three compliance programs. All have been revised since their initial publication, but still there remain differences in verbiage, and in some cases content, among the FDA, EPA, and OECD GLP principles, but the result for each is the assurance that the experimental information generated under each program is of sufficient quality and integrity to support the reports for the various studies.”
GxP in all of its forms
The acronym GLP, or “Good Laboratory Practices,” follows from another acronym, GxP. The latter encompasses a broad range of compliance-related activities or sets of regulations and quality guidelines developed to ensure the safety of life sciences products such as food and medical products, including drugs and medical devices and software applications, while maintaining the quality of processes throughout all stages of manufacturing, control, storage, and distribution. The variable “x” depends on the application of the standards. It can be M for “manufacturing,” C for “clinical,” L for “laboratory,” S for “storage,” D for “distribution,” R for “review,” etc. The GxP guidelines’ purposes are to ensure that regulated organizations follow standard processes.
For example, with Good Manufacturing Practices (GMP), the FDA regulates the quality of products such as drugs, medical devices, and active pharmaceutical ingredients by monitoring drug manufacturers' compliance with its Current Good Manufacturing Practices (CGMP). According to the FDA, all of these regulations “contain minimum requirements for the methods, facilities, and controls used in manufacturing, processing, and packing of a drug product. The regulations make sure that a product is safe for use and that it has the ingredients and strength it claims to have.”
Assessors and investigators for the FDA include a review of the manufacturer’s compliance with the CGMPs with any approval of a new or generic drug marketing application. This review shows that the FDA has determined that the company has the necessary facilities, equipment, and ability to manufacture the drug it intends to market.
Good Distribution Practices (GDP) ensure that when medical products arrive in the hands of patients, both suppliers and customers can be confident these products are effective, unadulterated and safe to use.
Good Review Practice (GRP) is a “documented best practice” related to the process, format, content, and management of a product review within the Center for Drug Evaluation and Research (CDER). GRPs are developed over time to improve the quality of reviews and provide consistency, clarity, and transparency to the overall review process of new products.
These documented best practices frequently change because of new science, statutes, regulations, guidance, and accumulated experience. As such, FDA staff must regularly update their policies and adopt them into their daily review activities. The most up-to-date documents can be found on the FDA Guidance Documents, and CDER Manual of Policies & Procedures (MAPP) pages.
Sorting through the confusion about GMPs and GLPs
The differences between GMPs and GLPs are important for those who must apply them or collaborate with those who do, especially in the lab.
Whereas GLPs are based on study, GMPs are concerned with process. GLPs focus on preclinical development, such as nonclinical laboratory safety studies that support or are intended to support the research or marketing applications of the product, including biocompatibility, toxicology, pharmacology, etc., while GMPs demonstrate to the FDA whether certain quantities of regulated products are manufactured according to predefined criteria.
GLPs only apply to studies performed in test systems like live organisms, plants, and microorganisms. Their purpose is to give the FDA the information to scrutinize and audit the scientific validity of research studies. The GMPs, on the other hand, should demonstrate to the FDA that batches of regulated products are manufactured according to predefined criteria. “Lot release or lot conformance testing for products on the market are almost always required to be done under GMP while testing of safety and efficacy should be done under GLP in most cases,” The FDA Group states.
Bailey MacDougall writes “Generally, most nonclinical studies will fall under the GLP regulation requirements. However, there are some instances or stages in the early pre-clinical phase where adherence to GLP is not required. Examples of studies that could be exempt from GLP include exploratory genotoxicity, mutagenicity, safety pharmacology, and general in-vitro toxicology studies.”
DotCompliance, a quality and compliance regulation company, states that knowing the differences between GMP and GLP can help differentiate between protocols needed during various production phases, as GMP and GLP affect different stages in the processes.
“GLP compliance monitors conditions, processes, documentation and archiving of studies performed in laboratory settings. Such endeavors require highly accurate and trackable data as well as high-quality results to validate and market the final product. GMP compliance refers to quality assurance in the manufacturing processes, as defined by the local regulatory body to ensure the safety of healthcare products sold in their jurisdiction.”
CGMP outlines the pre-approved quality requirements and specific conditions for manufacturing, such as lighting, plumbing, hygiene, storage, equipment maintenance, and separation of substances to avoid contamination. CGMP requires manufacturers of medications to obtain appropriate quality raw materials, establish robust operating procedures, detect and investigate product quality deviations, and maintain reliable testing laboratories. “This helps to prevent instances of contamination, mix-ups, deviations, failures, and errors.”
Requirements were made flexible so that each manufacturer could choose how to best implement controls using scientifically sound design, processing methods, and testing procedures. As the "C" in CGMP stands for "current," companies must use the latest technologies and systems to follow regulations. For example, equipment that may have been "top-of-the-line" to prevent contamination, mix-ups, and errors 20 years ago may be less than adequate by today's standards.
The FDA reminds its readers that CGMPs are minimum requirements. “Many pharmaceutical manufacturers are already implementing comprehensive, modern quality systems and risk management approaches that exceed these minimum standards.”
How to get ahead of the game
Here are some ways to avoid common laboratory practice errors in both environments:
- Be aware of all labels and signs, including caution and warning signs and any labels on equipment or materials.
- Keep work areas and yourself clean. Wash your hands, and don’t wear your lab coat outside the lab. Use approved cleaning procedures and solutions on surfaces. Check pest control devices regularly.
- Don’t use expired materials.
- Fill out all appropriate forms, and fill them in entirely, even if you’re just writing N/A. Always enter information immediately and sign your name and the date on any information you enter using black, indelible ink. Initial corrections, and never backdate or falsify information. Record all identifying factors, including part, lot, document, revision, and other control numbers. Have someone else double-check your work if you have to.
- Always report mistakes, even if you just suspect a mistake, to the appropriate supervisor.
- Never rely on first-time measurements.
- Always run a control sample.
Adhering to government guidelines can be complicated and is often confusing. Following these general ground rules could save enormous time and grief.